SÍNDROME DE EHLERS-DANLOS TIPO HIPERMOVIL: CONCORDANCIA DE LOS CRITERIOS DE DIAGNÓSTICO DE LA CLASIFICACIÓN INTERNACIONAL 2017 CON LOS CRITERIOS DE BRIGHTON.
Hugo Villarroel-Ábrego.
Médico internista, cardiólogo y ecocardiografista, Fellow SIAC y Fellow ECOSIAC, Hospital de Diagnóstico Escalón, San Salvador, El Salvador.
INTRODUCCIÓN. El diagnóstico del síndrome de Ehlers-Danlos hipermóvil es eminentemente clínico y no existen pruebas genéticas específicas para su confirmación. Para hacer un diagnóstico firme, hasta que aparecieron en 2017 los nuevos lineamientos de la Clasificación Internacional de Síndromes de Ehlers-Danlos, se requería de los llamados criterios mayores y menores de Brighton; al comparar estos con los recién publicados está claro que los requisitos para el diagnóstico son ahora mucho más estrictos. OBJETIVO. Buscar la concordancia entre ambos criterios de diagnóstico en pacientes ya diagnosticados con síndrome de Ehlers-Danlos hipermóvil según criterios de Brighton. MÉTODOS. A todos los pacientes ya diagnosticados en una clínica de cardiología entre el 1 de enero de 2013 y el 31 de marzo de 2017 se les aplicó una nueva herramienta diagnóstica, según los criterios de la Clasificación Internacional, se buscó la concordancia entre ambos criterios usando el índice kappa de Cohen. RESULTADOS. La población final fue de 75 pacientes; solo en 30 casos se pudo confirmar el diagnóstico (40%) con los nuevos criterios. El índice de kappa fue cercano a 0, demostrando total ausencia de concordancia. CONCLUSIONES. Los criterios de Brighton probablemente han sido demasiado inclusivos, con alta sensibilidad y baja especificidad; solo el 40% de los casos están respaldados por los nuevos criterios. Es mandatorio disponer de una prueba genética para diagnóstico, mientras tanto podría ser necesaria una periódica revisión de cualquier intento de diagnóstico basado en criterios puramente clínicos.
PALABRAS CLAVE: síndrome de Ehlers-Danlos. Criterios de Brighton. Clasificación Internacional de Síndromes de Ehlers-Danlos. Concordancia.
ABSTRACT.INTRODUCTION. The diagnosis of hypermobile Ehlers-Danlos syndrome is eminently clinical and there are no specific genetic tests for confirmation. To make a firm diagnosis, until the new guidelines of the International Classification of Syndromes of Ehlers-Danlos appeared in 2017, the so-called major and minor criteria of Brighton were required; When comparing these with the newly published ones it is clear that the requirements for diagnosis are now much stricter. OBJECTIVE. Search for agreement between both diagnostic criteria in patients already diagnosed with hypermobile Ehlers-Danlos syndrome according to Brighton criteria. METHODS All patients already diagnosed in a cardiology clinic between January 1, 2013 and March 31, 2017 were given a new diagnostic tool, according to the criteria of the International Classification, the agreement between both criteria was sought using the Cohen’s kappa index. RESULTS The final population was 75 patients; Only in 30 cases could the diagnosis be confirmed (40%) with the new criteria. The kappa index was close to 0, showing total absence of agreement. CONCLUSIONS The Brighton criteria have probably been too inclusive, with high sensitivity and low specificity; only 40% of cases are supported by the new criteria. It is mandatory to have a genetic test for diagnosis, meanwhile a periodic review of any diagnostic attempt based on purely clinical criteria may be necessary.
KEYWORDS: Ehlers-Danlos syndrome. Brighton criteria. International Classification of Ehlers-Danlos Syndromes. Agreement.
Las enfermedades hereditarias del tejido conectivo son consideradas por muchos médicos como rarezas clínicas. Sin embargo, el síndrome de hiperlaxitud articular, mejor llamado síndrome de Ehlers-Danlos tipo hipermóvil (SEDh, antes tipo III) es la menos rara de las enfermedades hereditarias de la colágena, aunque su prevalencia real es, sin embargo, desconocida. La colágena es una proteína fundamental del tejido conectivo y, si una mutación causa síntesis de colágena anormal, se reducen la resistencia y la integridad de la piel, articulaciones y otros tejidos. La hipermovilidad articular es una característica relevante en la mayoría de estos casos, pero alrededor del 4-10% de la población adulta saludable puede ser hipermóvil (1,2). Solo en una minoría de pacientes hiperlaxos hay una condición patológica y los criterios para distinguir a los hiperlaxos «sanos» de aquellos que deberían considerarse casos patológicos son la
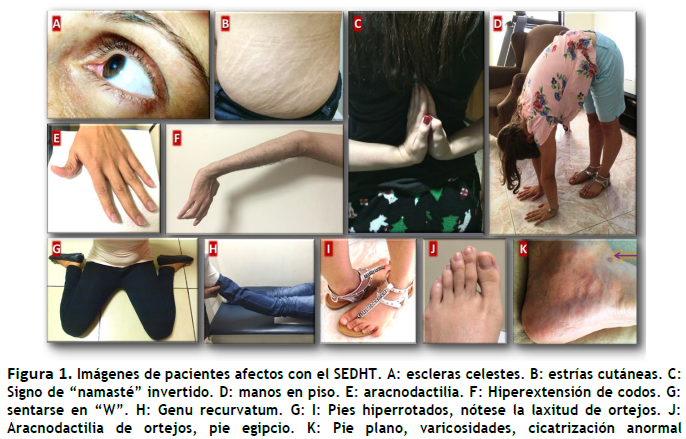
presencia de síntomas clínicos y signos de lesión tisular estructural, como ocurre en los síndromes de Ehlers-Danlos. La prevalencia varía entre las diversas razas, es mayor entre las mujeres y más notoria en los niños; aunque está presente desde el nacimiento, pero los síntomas pueden presentarse a cualquier edad. Esta no es una condición letal y permite una sobrevida normal, pero es potencialmente seria y no una condición leve: Es una enfermedad genética, autosómica dominante, que puede afectar muchos tejidos y dar mala calidad de vida. Algunas de las manifestaciones típicas del SEDh se muestran en la Figura 1. En general, estas enfermedades merecen un estudio exhaustivo de todos los órganos y sistemas para diagnóstico precoz e intervenciones terapéuticas oportunas; por otra parte, un diagnóstico diferencial acertado es necesario antes de que se indiquen pruebas genéticas específicas, que no siempre son necesarias y tienen un alto costo.
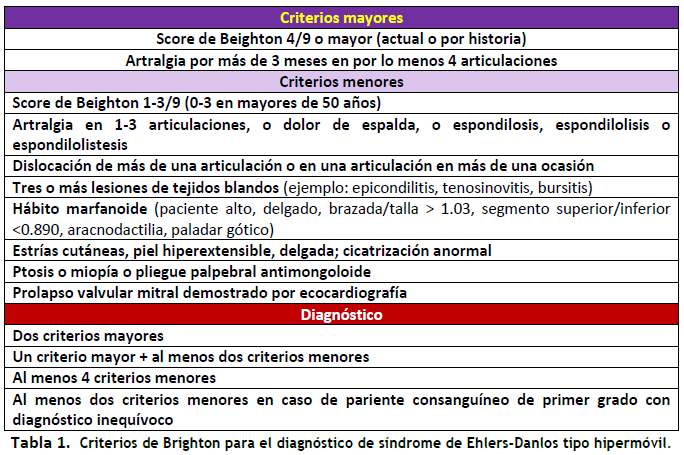
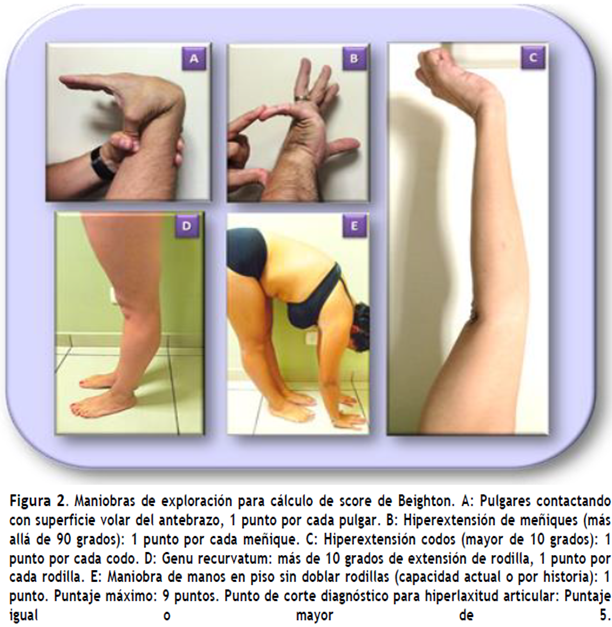
El diagnóstico de SEDh es eminentemente clínico y no hay pruebas genéticas específicas de confirmación porque se desconoce el gen (o genes) involucrados (1-3). Para realizar un diagnóstico firme, al menos hasta que las pautas de la Clasificación Internacional de los Síndromes de Ehlers-Danlos (CI-SED) fueran publicadas el año 2017 (4), se requirió la puntuación de movilidad articular de Beighton, (5) incorporada como uno de los criterios principales de Brighton (6) para el diagnóstico de SEDh (ver Figura 2 y Tabla 1).
Desde que se informaron muchas ambigüedades en la evaluación de casos de posible diagnóstico, considerando los once subtipos
propuestos en la nosología de Berlín de 1988, la nosología de Villefranche de 1998 (5) redujo el número de subtipos a solo seis, en un intento por actualizar los últimos descubrimientos en el campo de la genética con el perfil fenotípico respectivo de cada mutación; esto, definiendo para cada subtipo un conjunto de criterios diagnósticos mayores y menores. Veinte años después la genética continúa avanzando, tanto que ha sido posible la detección de nuevos subtipos y, considerando que el SEDh todavía no se ha vinculado a una mutación específica, Malfait F, Francomano C, Byers P et al diseñaron una nueva clasificación general y, para los casos de SEDh, una serie de criterios obligatorios, más restrictivos.
Los tres nuevos criterios para el diagnóstico de SEDh (6) son:
CRITERIO 1: Hipermovilidad articular generalizada.
- Beighton score. Punto de corte: 5 o más puntos de 9 posibles; para adultos mayores de 50 años se ha propuesto >4 puntos y para niños (prepuberales) y adolescentes >6 puntos.
- Cuestionario de 5 puntos. Dos o más respuestas positivas tienen alta sensibilidad y especificidad para diagnóstico de hipermovilidad articular.
- ¿Puede (o alguna vez pudo) poner las manos en el piso sin doblar las rodillas?
- ¿Puede (o alguna vez pudo) doblar los pulgares hasta tocar el antebrazo?
- ¿De niño le gustaba contorsionar su cuerpo de manera extraña o podía hacer el “split”?
- ¿De niño o adolescente se le dislocó un hombro o la rótula en más de una ocasión?
- ¿Se considera usted hipermóvil o muy flexible o elástico?
CRITERIO 2: Dos o más de los tres grupos de características tipificadas como A, B y C: (A+B, A+C, B+C, A+B+C)
- Características A: Manifestaciones sistémicas de un desorden más generalizado del tejido conectivo). Deben estar presentes al menos 5 condiciones:
- Piel inusualmente suave y/o aterciopelada.
- Leve hiperextensibilidad cutánea (> 1.5 cm).
- Estrías cutáneas no explicadas.
- Pápulas piezogénicas en talones.
- Hernias abdominales múltiples o recurrentes.
- Cicatrices atróficas en al menos dos sitios sin aspecto papiráceo o hemosiderótico.
- Prolapso del piso pélvico, rectal y/o uterino sin historia de factores predisponentes.
- Apiñamiento dental y/o paladar abovedado/estrecho.
- Aracnodactilia: Signo de Steinberg positivo (muñeca) bilateral y/o signo de Walker positivo (pulgar) bilateral.
- Relación brazada/talla > 1.05.
- Prolapso valvular mitral.
- Dilatación de raíz aórtica con Z-score > +2.
- Características B: Historia familiar positiva en parientes de primer grado que independientemente llenan los criterios de SEDh.
- Características C: Deben llenarse al menos uno de los siguientes requisitos:
- Dolor musculoesquelético en dos o más miembros con recurrencia diaria al menos por 3 meses.
- Dolor crónico generalizado por más de 3 meses.
- Luxación recurrente de articulaciones o franca
-
inestabilidad articular en ausencia de trauma:
- Tres o más luxaciones atraumáticas en la misma articulación o dos luxaciones atraumáticas en dos articulaciones diferentes en distintos momentos.
- Confirmación médica de inestabilidad de al menos dos articulaciones, en ausencia de trauma.
CRITERIO 3. Deben llenarse todos los siguientes requisitos:
- Ausencia de excesiva fragilidad cutánea que haga sospechar otras variantes del SED.
- Exclusión de otras enfermedades adquiridas o heredables del tejido conectivo, incluyendo condiciones reumatológicas autoinmunes.
- Exclusión de diagnósticos alternativos que puedan incluir hiperlaxitud debida a hipotonía y/o laxitud del tejido conectivo.
El objetivo de la presente investigación es buscar concordancia entre los criterios de Brighton y los nuevos criterios de la Cl-SED, en una población de 75 pacientes ya diagnosticados con SEDh.
MATERIALES Y MÉTODOS.Se utilizó un diseño de concordancia de dos escalas diagnósticas (Brighton vs CI-SEDh) en una serie de 75 pacientes con diagnóstico previo de SEDh por criterios de Brighton, entre 2013 y 2017, en una clínica ambulatoria de cardiología en San Salvador, El Salvador. Criterios de inclusión:
- Consultante en clínica general de cardiología.
- Edad mayor o igual a 12 años, pero menor de 50 años.
- Diagnóstico confirmado de SEDh según los criterios de Brighton: 2 criterios principales, 1 criterio principal asociado con dos criterios menores, cuatro criterios menores y dos criterios menores en el caso de parientes consanguíneos con un diagnóstico inequívoco. Criterios de exclusión:
- Edad menor de 12 años o mayor de 50 años.
- Pacientes con diagnósticos confirmados de osteogénesis imperfecta, síndromes de Marfan, Shprintzen-Goldberg o Loeys-Dietz o cualquiera de los otros tipos de síndrome de Ehlers-Danlos diferentes del tipo hipermóvil.
- Enfermedades autoinmunes del colágeno.
- Familiares asintomáticos de pacientes seleccionados.
- Datos de historia clínica.
- Fecha de nacimiento, edad y sexo.
- Historia de anomalías reumatológicas y ortopédicas. Antecedentes de lesiones ligamentosas, articulares, esqueléticas o de tejidos blandos.
- Signos vitales y antropometría.
- Peso, talla, amplitud de brazada, relación brazada/talla.
- Presión arterial y frecuencia cardíaca.
- Examen físico.
- Facies y aspecto general.
- Revisión de ojos, oídos, nariz, cavidad oral.
- Revisión de piel, faneras y tejidos blandos.
- Exploración integral del aparato cardiovascular.
- Score Beighton de hipermovilidad articular: meñiques, pulgares, codos, rodillas y maniobra de
- manos en el piso. Puntaje posible: 0-9.
- Procedimientos auxiliares de diagnóstico:
- Electrocardiograma
- Ecocardiograma doppler color.
Todos los archivos e instrumentos recopilados fueron revisados y los nuevos criterios de CI-SED se aplicaron, buscando la concordancia global del diagnóstico. Análisis estadístico: La base de datos fue procesada en software estadístico para calcular el índice kappa de Cohen, el cual fue interpretado de la siguiente manera (Landis y Koch):
- < 0.20: Pobre concordancia.
- 0.20 a 0.40: Débil concordancia.
- 0.40 a 0.60: Moderada concordancia.
- 0.60 a 0.80: Buena concordancia.
- > 0.80: Muy buena concordancia.
Se disponía de una casuística de 75 pacientes con SEDh, diagnosticados entre 2013 y 2017 según los criterios de Brighton, con edad promedio de 29+11.2 años, la mayoría de sexo femenino n=54, 75%). Las características clínicas de los 72 pacientes están detalladas en la Tabla 2. De los 75 casos estudiados se diagnosticó SEDh en tan solo 30 pacientes según los nuevos criterios (40%). Se calculó el índice kappa, obteniéndose un valor de 0 con error estándar 0.094 (intervalo de confianza -0.185 a 0.185), encontrándose por tanto una pobre concordancia entre ambas escalas diagnósticas.

El diagnóstico de SEDh no puede basarse en una prueba genética, al menos hasta la fecha. Esto requiere que los criterios clínicos se optimicen regularmente, de modo que sin ser demasiado inclusivos (como los de Brighton), tampoco se caiga en el caso de perder excesiva sensibilidad a expensas de una muy elevada especificidad. Muchos pacientes con cuadro clínico altamente sospechoso pero que no llenan los requisitos exigidos por la Clasificación Internacional podrían, sin embargo, ser reconsiderados para el diagnóstico de SEDh en futuras revisiones. Al no haber patrón oro para diagnóstico estos criterios seguirán siendo recomendaciones de expertos, el nivel de evidencia menos poderoso. Es evidente que hay muy pobre concordancia entre ambas estrategias para diagnosticar SEDh y eso obliga a considerar las opciones diagnóstica y de manejo para todos los casos (60% del total) que no califican para la CI-SEDh. Estos pacientes tienen síntomas y discapacidades y deben ser atendidos independientemente si se pueden etiquetar o no como portadores del síndrome; el seguimiento es indispensable porque el fenotipo del paciente tiende a cambiar con la edad. Por ejemplo, el envejecimiento conduce a una pérdida natural de la elasticidad de las articulaciones; de ahí la necesidad de reducir el valor de corte a 4 puntos en personas mayores de 50 años; además hay pacientes que, debido a un dolor intenso o lesiones incapacitantes, no pueden realizar las maniobras adecuadas para el diagnóstico. Otras consideraciones muy relevantes respecto al SEDh y que deben ser tomadas en cuenta a futuro, a la hora de reformular criterios de diagnóstico, incluyen:
- Es muy probable que el diagnóstico no se pueda hacer transversalmente en pacientes muy jóvenes, en los que aún no predominan los síntomas musculoesqueléticos. Por otro lado, en niños y adolescentes es normal que exista un cierto grado de hiperlaxitud articular. El seguimiento de algunos de estos casos es de gran importancia, en especial si uno o ambos padres tiene diagnosticada la condición.
- Después de múltiples episodios de dislocaciones, algunos pacientes han perdido elasticidad.
- El score de Beighton no considera la hiperlaxitud de muchas otras articulaciones y la caída del arco del pie (pie plano), por el momento no hay solución satisfactoria para estas limitantes según los expertos redactores de la CI-SEDh.
- Debería reconsiderarse la importancia de los signos oculares, descartados en la CI-SEDh. La prevalencia de la miopía varía en las diferentes poblaciones del mundo y la incidencia aumenta de manera acelerada. Al ser un problema tan generalizado, es difícil atribuirle importancia como criterio de diagnóstico de SEDh; sin embargo, la mayoría de los pacientes tiene anomalías oculares, a menudo graves, como miopía patológica, queratocono, xeroftalmía, anomalías vítreas y opacidades corneales. Quizás las alteraciones importantes de la esfericidad corneal y la miopía patológica (más de -6,0 dioptrías) deben considerarse entre las características A del Criterio 2 de la CI-SEDh.
- Los criterios de Brighton probablemente han sido demasiado inclusivos, con alta sensibilidad y baja especificidad, por lo que solo el 40% de los casos son compatibles con los nuevos criterios.
- Todos los diagnósticos realizados por los criterios de Brighton deben ser revisados a la luz de los nuevos criterios CI-SED pero esto no implica que los pacientes que no llenan criterios deban ser cesados de cuidado médico: muchos tienen dolor, discapacidad, disautonomía o problemas oculares que requieren de atención especializada y seguimiento.
- Estos criterios se pueden reconsiderar en el futuro para alcanzar el máximo valor predictivo, al menos mientras se detecta la mutación (o mutaciones)
-
responsable de los SEDh, especialmente en el campo de la investigación clínica.
- El seguimiento de pacientes muy jóvenes sospechosos de SEDh que aún no cumplen con los criterios requiere un seguimiento a largo plazo, especialmente si hay parientes consanguíneos con un diagnóstico confirmado.
- Quizá un enfoque multidisciplinario sea ideal: médico de cabecera, ortopeda, oftalmólogo y cardiólogo deben involucrarse en equipo para el manejo integral de estos casos.
Financiamiento: El autor no recibió patrocinio para llevar a cabo este artículo.
Conflicto de intereses: El autor declara que no tiene conflictos de intereses.
- Bravo, JF. Síndrome de Ehlers-Danlos con especial énfasis en el síndrome de hiperlaxitud articular. Rev Med Chile 2009; 137: 1488-1497.
- Kirk JA, Ansell BM, Bywaters EGL. The hypermobility syndrome. Ann. Rheum. Dis. (1967), 26, 419.
- Castori M. Ehlers-Danlos Syndrome, Hypermobility Type: An underdiagnosed Hereditary Connective Tissue Disorder with Mucocutaneous, Articular and Systemic Manifestations. ISRN Dermatology Volume 2012 Article ID 751768.
- Malfait F, Francomano C, Byers P et al. The 2017 International
-
Classification of the Ehlers-Danlos Syndromes. Am J Med Genet Part C Semin Med Genet 175C:8-26.
- Beighton P, De Paeppe A, Steinmann B et al. Ehlers-Danlos nosology. Am J Med Genet. 1998 Apr 28;77(1):31-7.
- Grahame R. Brighton Diagnosis Criteria for the Benign Joint Hypermobility Syndrome. Br. J Rheumatol 2000; 27: 1777-1779.
- Tinkle B, Castori M, Berglund B et al. Hypermobile Ehlers-Danlos Syndrome (a.k.a. Ehlers-Danlos Syndrome Type III and Ehlers-Danlos Syndrome Hypermobility Type): Clinical Description and Natural History. Am J Med Genet Part C Semin Med Genet 175C:48-69.