FACTORES DE RIESGO DE MORTALIDAD EN PACIENTES CON LINFOHISTIOCITOSIS HEMOFAGOCITICA REACTIVA.
Factores de Riesgo de Mortalidad en Pacientes con Linfohistiocitosis Hemofagocitica Reactiva.
RESUMEN
La Linfohistiocitosis Hemofagocítica (LHH) es una condición caracterizada por fiebre, pancitopenia, hepatoesplenomegalia, evidencia de proliferación histiocítica con actividad fagocítica en la medula ósea y otros órganos del sistema retículo endotelial. Los siguientes son factores descritos que influencian la morbilidad/mortalidad: plaquetopenia, aumento de la ferritina, coagulación intravascular diseminada, malignidad hematológica, fiebre, aumento de LDH, edad, presencia de visceromegalia, aumento de la bilirrubina, elevación marcada de las transaminasas, comorbilidad, falta de respuesta al tratamiento e hipocelularidad en la medula ósea inicial.
Materiales y métodos:
se realizó un estudio observacional analítico de casos y controles en pacientes diagnosticados con LHH en el Hospital Nacional Rosales, obtenidos del registro durante el periodo entre 2010 y 2015, que cumplían con los criterios de diagnóstico propuestos por Henter y col. comprobación de hipótesis mediante la prueba Chi2 y ejecución de análisis univariado y multivariado de las variables.
Resultados:
se incluyeron 101 casos de LHH reactiva, 40 pacientes registrados como fallecidos conformaron el grupo caso. Los factores de riesgo investigados plaquetopenia, aumento de la ferritina, coagulación intravascular diseminada, malignidad hematológica, fiebre, aumento de LDH, edad, presencia de visceromegalia, aumento de la bilirrubina, elevación marcada de las transaminasas, comorbilidad, falta de respuesta al tratamiento e hipocelularidad en la medula ósea inicial mostraron relacion con mortalidad estadísticamente significativa.
Conclusión:
La LHH reactiva tiene una alta letalidad, y en nuestra serie, se ratificaron los factores de riesgo ya descritos en la literatura internacional.
Palabras claves: Linfohistiocitosis hemofagocitica, mortalidad, factores de riesgo.
ABSTRACT.
Hemophagocytic lymphohistiocyosis síndrome (HLH) is a condition characterized by fever, pancytopenia, hepatoesplenomegaly, evidence of histiocytic proliferation with phagocytic activity in bone marrow and other organs from the reticuloendothelial system. The following have been identified as risk factors for mortality: thrombocytopenia, increased seric ferritin levels, intravascular coagulopathy, hematological malignancy, fever, increased DHL levels, age, presence of visceromegaly, increased bilirrubin levels, increased transaminase levels, presence of co morbidities, lack of response to treatment and hypocelularity in the initial bone marrow aspiration.
Materials and methods.
An analytic observational case control study was conducted, with HLH patient’s files diagnosed and treated at Hospital Nacional Rosales, between 2010 and 2015, with 5 or more Henter et al criteria. Variables were taken to test literature proved risk factors and those factors suspected in the Ward. Hypothesis were tested with univariate and multivariate analysis.
Results
101 patients were included, with 40 conforming case group. Most of risk factors studied: thrombocytopenia, increased seric ferritin levels, intravascular coagulopathy, hematological malignancy, fever, increased DHL levels, presence of visceromegaly, increased bilirrubin levels, lack of response to treatment and hypocelularity in the initial bone marrow aspiration were ratified as related to mortality, but increased transaminase levels, presence of co morbidities, age above 60 years, and increased ferritin levels were not.
Conclusion .
HLH syndrome has a high rate of mortality and risk factors related to mortality described in the literature were ratified in our series.
Key words: Hemophagocytic lymphohistiocyosis, mortality, risk factors.
INTRODUCCION.
La Linfohistiocitosis Hemofagocítica (LHH) o Síndrome Hemofagocítico es una condición caracterizada por fiebre, hepatoesplenomegalia, pancitopenia, proliferación histiocítica con evidencia de actividad fagocítica en la medula ósea y/o en otros órganos del sistema retículo endotelial como resultado de la activación patológica del sistema inmune con signos y síntomas de inflamación excesiva, resultado de la disfunción de las células natural killer (NK) que conlleva a sobreestimulación, proliferación y migración de células T. Fue descrita por primera vez en 1939 bajo el designio de “Reticulosis Medular Histiocítica”1 como una variante atípica de linfomas de Hodgkin y para el año de 1952 se identifica como un desorden genético denominado “Reticulosis Familiar Hemofagocítica”2. La incidencia se estima en 1.2 casos/millón de individuos/año sin embargo se infiere que este dato es subestimado pues en muchas ocasiones no se sospecha el diagnostico3. Otras de las principales manifestaciones clínicas del síndrome son esplenomegalia, hipertrigliceridemia y/o hipofibrinogenemia.
Clasificación
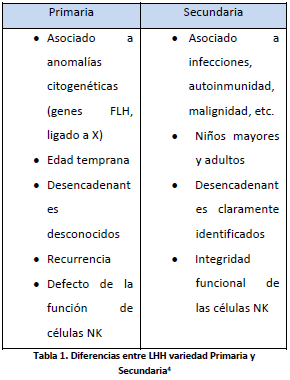
Tiene 2 formas de presentación: la hereditaria (primaria) y la adquirida (secundaria o reactiva). Las diferencias de ambas podemos verlas en la tabla 14.
Linfohistiocitosis Hemofagocítica Secundaria o Reactiva.
Esta variedad se asocia a causas infecciosas, neoplasias (principalmente enfermedades linfoproliferativas), enfermedades autoinmunes y algunas enfermedades metabólicas. El virus Epstein-Barr es el más frecuentemente
asociado, seguido por virus (citomegalovirus, hepatitis A, B y C, herpes simple, inmunodeficiencia humana, etc), bacterias (micobacterias, micoplasmas, ricketsias, borrelia, treponemas, etc), hongos (cándida, histoplasma, criptococo, aspergillus, etc) y parásitos (babesia, leishmania, plasmodium, etc). También se ha asociado a enfermedades malignas, principalmente a leucemias o linfomas de células T / NK, y en linfomas anaplásicos, linfomas difusos de células grandes, leucemias linfoblásticas B, leucemias mieloides y con menor frecuencia en tumores germinales y otros tumores sólidos6. En muchos casos coexiste en forma simultánea estados infecciosos en el contexto de un sistema inmune funcionalmente alterado por efecto de la quimioterapia o por la producción de citosinas tumorales. En el contexto de las enfermedades autoinmunes la condición es conocida como Síndrome de Activación de Macrófagos. Es una complicación propia de la artritis reumatoidea juvenil, aunque también puede presentarse en otras enfermedades. Las manifestaciones incluyen fiebre, visceromegalia, linfadenopatía y coagulación intravascular diseminada.
La presencia de citopenias es un hallazgo tardío. Los pacientes presentan una disminución en la actividad de las células NK, disminución de la expresión de perforina y altos niveles de CD5 y CD163. Generalmente mantiene una buena respuesta al tratamiento con inmunosupresores y dosis altas de inmunoglobulina7.
Diagnóstico
En la variedad adquirida o secundaria el diagnostico se establece con la presencia de 5 de 8 criterios diagnósticos propuestos por Henter y cols9. y aceptados por la sociedad del histiocito, que son los siguientes:
Criterios de Diagnostico Inicial:
- Fiebre
- Esplenomegalia
- Citopenias (comprometiendo al menos 2 de las 3 líneas celulares (Anemia con Hb < 9 gr%, Plaquetas < 100,000 /mm3, Neutrofilos < 1,000/ mm3
- Hipertrigliceridemia y/o hipofibrinogenemia: Trigliceridos en ayuno > 265 mg%, Fibrinogeno < 150 mg%.
- Evidencia de hemofagocitosis en medula ósea, bazo o ganglios linfáticos.
Nuevos criterios de diagnóstico:
- Nivel de actividad de células NK bajo o ausente
- Nivel de ferritina > 500 μg/L
Comentarios.
- Si la actividad hemofagocitica no se encuentra en el momento de presentación, se debe buscar posteriormente. Si la medula ósea no es concluyente se debe buscar en otros órganos. La realización de múltiples aspirados de MO pueden ser requeridos para confirmar el diagnostico.
- Los siguientes hallazgos pueden proveer una fuerte evidencia de soporte para diagnostico: (a) presencia de pleocitosis, por presencia de células mononucleares y/o elevación de las proteínas del líquido cerebro-espinal (b) Histopatología hepática con evidencia de hepatitis crónica persistente.
- Presencia de otros hallazgos clínicos o de laboratorio que son consistentes con el diagnóstico: síntomas meningo-encefaliticos, adenomegalias, ictericia, edema, rash cutáneo, alteración de las enzimas hepáticas, hipoproteinemia, hiponatremia, VLDL alta y HDL baja9.
La fiebre y la esplenomegalia son las manifestaciones más constantes, presentes en hasta el 75% de casos mientras que la bicitopenia, hipertrigliceridemia y ferritina > 500 ng/ml se encuentran en la mitad de los casos. La evidencia del fenómeno hemofagocítico morfológico es variable y su prevalencia va del 25 al 100% sin embargo es un hallazgo en otras condiciones tales como infecciones, enfermedades autoinmunes, reacciones transfusionales. Los niveles elevados del receptor soluble de IL-2 y las alteraciones funcionales de las células NK se encuentran elevados en > 90% de casos10. Se manifiesta además una variedad de hallazgos dermatológicos:
rash maculopapular, morbiliforme, eritema, paniculitis, etc11. Los que desarrollan compromiso pulmonar tienen peor pronóstico. Hasta un tercio de pacientes se manifiesta con síntomas neurológicos y en hasta el 50% de los casos en líquido cerebro-espinal es anormal, la neuropatía periférica difusa, secundaria a destrucción de mielina por macrófagos se presenta hasta en 70% de los casos12.
FACTORES PRONÓSTICOS
Al momento existe una serie de estudios clínicos para sustentar una escala de severidad de la enfermedad. Los siguientes son factores de riesgo que influyen en la mortalidad de los pacientes con síndrome LHH, en orden de importancia; la presencia de plaquetopenia de < 100,000/mm3 13,14,15,17,18,20,21,24,25,28, aumento de la ferritina > 500 μg/dL 16,20,23,24,25,27, presencia de coagulación intravascular diseminada con fibrinógeno < 100 mg/dL o prolongación de aTTP/TP > 1,5 veces el control 13,18,20,23,24, malignidad hematológica: leucemia o linfoma de
células B o T/NK 14,16,19,22, fiebre > 38,3 °C 13,26,28, aumento de LDH > 225 UI/L 14,18,24, edad > 60 años 14,15, presencia de visceromegalia (hepatoesplenomegalia por ecografía o clínica)15,17 y aumento de la bilirrubina total > 2 mg/dL 13,25.
En la experiencia del servicio de Hemato-Oncología del Hospital Nacional Rosales a cargo del Dr. José Héctor Valencia, se han observado otros factores a considerar y que no se mencionan en la literatura actual: elevación marcada de las transaminasas (TGO/TGP > 1,000 UI/L), Comorbilidades (DM2, Cardiopatías), Falta de respuesta al tratamiento con protocolo HLH-2004 evidenciado por persistencia de la fiebre después de 2 semanas y/o persistencia de la actividad hemofagocítica en la MO, Hipocelularidad en la MO inicial con fenómeno hemofagocítico y/o progresión hacia aplasia medular.
En vista de la alta frecuencia de este síndrome en el Servicio de hematooncologia y de la alta mortalidad, se decidió realizar el presente estudio con el objetivo primario de identificar los factores relacionados a mortalidad en el Síndrome LHH.
MATERIALES Y METODOS.
Se realizo un estudio Observacional Analítico de Casos y Controles de los factores de riesgo de mortalidad en los pacientes diagnosticados con Linfohistiocitosis hemofagocítica reactiva en el Hospital Nacional Rosales. Se realizo utilizando fuentes documentales retrospectivas, los expedientes de los pacientes que consultaron y fueron tratados en el HNR. La Población del estudio se definió:
Caso: al paciente con LHH reactiva, que falleció durante el evento
Control:paciente con LHH reactiva que no falleció durante el evento.
No se hizo apareo (matching) debido a que el número de pacientes con LHH al momento es limitado, se incluirán todos los pacientes según sus características con respecto al grupo caso o control y se incluirían a todos los pacientes identificados.
CRITERIOS DE INCLUSIÓN
- Diagnóstico de LHH confirmado por 5 criterios diagnósticos y que se encuentra plasmado en el expediente.
- Diagnosticados y manejados en el HNR.
- Expedientes existentes y completos.
CRITERIO DE EXCLUSIÓN
- Expediente perdido.
- Ingresado para tratamiento y paciente se retira o fallece sin confirmación del diagnóstico.
Una vez identificados en el registro de Hemato-oncología los pacientes con diagnostico de LHH, se revisaron los expedientes para tomar las variables necesarias para el estudio: edad, sexo, procedencia, cumplir con 5 o más criterios de Henter et al, presentación clínica, factores de riesgo presentes: plaquetopenia, aumento de la ferritina, presencia de CID, fiebre, malignidad hematológica, edad, visceromegalia, valor de LDH, valor de transaminasas, falta de respuesta al tratamiento, medula ósea inicial hipocelular, comorbilidad, valor de bilirrubina total; tratamiento recibido y muerte.
Análisis estadístico. Se hizo estadística descriptiva de las características del grupo caso y del grupo control. Luego se
realizaron pruebas de comprobación de hipótesis de los factores de riesgo planteados en hipótesis nulas. Se trabajo con un valor de nivel de significancia estadística de 0.05 y se hizo análisis univariado y multivariado de los factores.
RESULTADOS.
En el registro de Hemato-oncología HNR se encontraban anotados 207 pacientes, de los cuales 101 cumplían con 5 o más criterios de Henter y criterios de inclusión que fueron los que conformaron nuestro grupo de estudio. Observando a los pacientes excluidos (104) se encontró una tasa de mortalidad de 8.65% (9 de 104), mientras que en el grupo incluido con diagnostico confirmado de LHH, se encontró una tasa de mortalidad de 39.60% (40 de 101), con una asociación estadísticamente significativa entre tener el diagnostico de LHH por criterios de Henter y mortalidad (p= 0.000).
Características socio demográficas.
Las características socios demográficos y clínicos de ambos grupos, podemos verlas en la tabla 1.
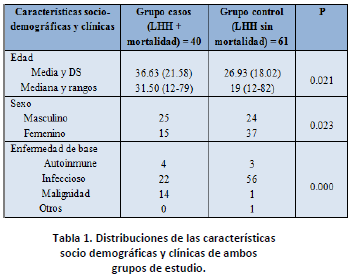
Observamos que hay diferencias en las características para ambos grupos.
Factores de riesgo de mortalidad.
En cuanto a las variables investigadas como factores de riesgo y su relación con la mortalidad se obtuvo lo siguiente tanto en análisis univariado, como en análisis multivariado, observando que hiperferritinemia >500, elevación de transaminasas, edad mayor de 60 años, no mostraron significancia estadística, como factores de riesgo de mortalidad. Ver tabla 2
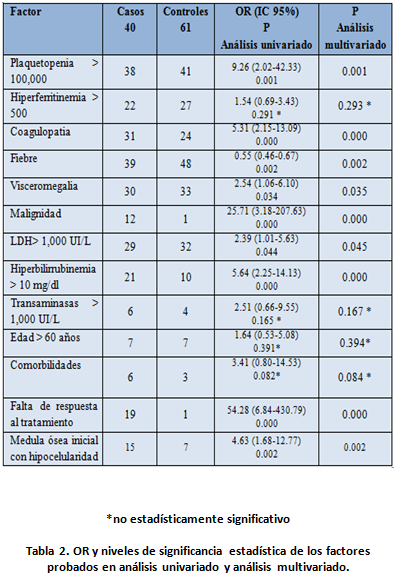
En nuestra serie se encontró una mortalidad del 39.60%, y una falta de respuesta al tratamiento del 19.80% en general, pero siendo el 95% de los casos en el grupo que falleció. Se reevalúo la relación entre los factores de riesgo de muerte categorizándolos por falta de respuesta al tratamiento encontrando una relación entre: plaquetopenia (p=0.009), visceromegalia (p=0.001), malignidad de base (p=0.011), LDH (p= 0.046), Bilirrubina > 10 mg/dl (p= 0.009),
presencia de comorbilidades (p= 0.000) y medula ósea hipocelular al inicio (p=0.005).
Tratamiento. El tratamiento que fue iniciado para los casos de LHH reactiva con más frecuencia era el que combinaba Etoposido a dosis de 150 mg/m2 dos veces por semana, Dexametasona 40 mg/día y Ciclosporina A 200 mg/día; en forma excepcional se encontró la implementación de plasmaféresis como tratamiento adyuvante al esquema descrito (n=1) y en aquellos pacientes que además de LHH reactiva se documentó malignidad recibieron protocolos de quimioterapia sistémica para respectiva condición basal (n=20, esquemas CHOP, MINE, BFM, Inducción 7/3).
DISCUSION. El objetivo primario del estudio era el de Determinar los factores de riesgo de mortalidad en pacientes diagnosticados con Linfohistiocitosis Hemofagocítica Reactiva en el Hospital Nacional Rosales. Creo haber alcanzado mi objetivo de forma parcial, ya que esta consciente que el tamaño muestral de los casos existentes podría estar limitando
En la serie identificada con diagnostico de egreso de LHH, solo el 48.79% cumplían el criterio diagnostico que consiste en tener de 5 a 8 criterios de Henter y col, lo que puede ser explicado por el hecho que fue catalogado como sospechoso de LHH y aunque no fue confirmado, quedo establecido como tal y censado así.
Se confirmo la relación entre la presentación de los factores de riesgo ya descritos por la literatura y la mortalidad en nuestra serie, tales como la presencia de plaquetopenia de < 100,000/mm3 13,14,15,17,18,20,21,24,25,28, presencia de coagulación intravascular diseminada 13,18,20,23,24, malignidad hematológica: leucemia o linfoma de células B o T/NK 14,16,19,22, aumento de LDH > 225 UI/L 14,18,24, presencia de visceromegalia (hepatoesplenomegalia por ecografía o clínica)15,17, tanto en el análisis univariado como en el análisis multivariado. Pero a diferencia de lo descrito en la literatura no se encontró relación estadísticamente significativa con edad mayor de 60 años, lo cual
puede haber sido probable por el bajo número de pacientes con edades avanzadas, ya que tuvimos una media de edades entre los 25 y 35 años. Tampoco encontramos relación con Hiperferritinemia. Llama la atención que en nuestra serie, se encontró un OR de 0.552 para fiebre (p=0.002), dando la impresión que la fiebre en nuestro caso fue más bien un factor protector de muerte y no un factor de riesgo. Debería de investigarse más este hallazgo en futuros estudios con tamaños de muestra más grande.
Por efecto propios del estudio, en lugar de utilizar el valor de corte para la hiperbilirrubinemia de 2 mg/dl13,25. Se tomó el valor de 10 mg/dl, y encontramos una relación estadísticamente significativa.
Con los factores de riesgo, propuestos por el servicio de Hematología: presencia de comorbilidades como diabetes mellitus, o cardiopatías, falla al tratamiento, hipocelularidad en la medula ósea inicial y elevación de las transaminasas, solo en falla al tratamiento e hipocelularidad en la medula ósea inicial estuvieron
relacionadas con mortalidad. Ya que el 95% de los pacientes que fallaron al tratamiento fueron los que fallecieron, sugerimos que también se debería realizar un estudio enfocado hacia factores de riesgo de falla terapéutica que sería similar a mortalidad.
REFERENCIAS.
- Scott R, Robb-Smith A. Histiocytic medullary reticulosis. Lancet. 1939; 2: 194-8.
- Farquhar JW, Claireaux AE. Familial haemophagocytic reticulosis. Arch Dis Child. 1952; 27: 519-25.
- Henter JI, Elinder G, Soder O, Ost A. Incidence in Sweden and clinical features of familial hemophagocytic lymphohistiocytosis. Acta Paediatr Scand. 1991; 80: 428-35.
- Risma K, Jordan MB. Hemophagocytic lymphohistiocytosis updates and envolving concepts. Curr Opin Pediatr 2012, 24; 1: 9-15.
- Allen M, De Fusco C, Legrand F, et al. Familial hemophagocytic lymphohistiocytosis: how late can the onset be? Haematologica. 2001; 86: 499-503.
- Janka GE, Imashuku S, Elinder G, et al. Infection and malignancy- associated hemophagocytic syndromes. Secondary hemophagocytic lymphohistiocytosis.Hematol Oncol Clin North Am. 1998; 12: 435-44
- Larroche C. Hemophagocytic lymphohistiocytosis in adults: Diagnosis and treatment. Joint Bone Spine 2012, 79; 4: 356-361.
- Woods CW, Bradshaw WT, Woods AG. Hemophagocytic lymphohisiocytosis in the premature neonate. Adv Neonatal Care 2009, 9; 6: 265-273.
- Henter JI, Horne A, Arico M, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatric Blood Cancer. 2007; 48: 124-31.
- Schram AM, Berliner N. How I Treat hemophagocytic lymphohistiocytosis in the adult patient. Blood 2015. 7, 125; 19: 2908-2914.
- Morrell DS, Pepping MA, Scott JP, Esterly NB, Drolet BA. Cutaneous manifestations of hemophagocytic lymphohistiocytosis. Arch Dermatol. 2002; 138(9): 1208-12.
- Thompson PA, Allen CE, Horton T, Jones JY, Vinks AA, McClain KL. Severe neurologic side effects in patients being treated for hemophagocytic lymphohistiocytosis.Pediatr Blood Cancer. 2009;52 (5): 621-5.
- Dao ATT, Luong VT, Nguyen TT, Huynh QTV, Phan TTT, Lam MT, Ngoma AKM and Koriyama Ch. Risk factors for early fatal outcomes among children with hemophagocytic lymphohistiocytosis (HLH): A single-institution case-series in Vietnam. Pediatr Hematol & Oncol 2014; 31: 271 – 281.
- Arca M, Fardet L, Galicier L, Riviere S, Marzac Ch, Aumont C, Lambotte O and Coppo P. Prognostic factors of early death in a cohort of 162 adults haemophagocytic syndrome: impact of triggering disease and early treatment with etoposide. Br J Haematol, 2015; 168: 63 – 68.
- Li J, Wang Q, Zheng W, Ma J, Zhang W, Wang W and Tian X. Hemophagocytic lymphohistiocytosis. Clinical analysis of 103 adult patients. Medicine, 2014; 93: 100 – 105.
- Rajagopala S, Singh N, Agarwal R, Gupta D and Das R. Severe hemophagocytic lymphohisiocytosis in adults-experience from an intensive care unit from north india. Ind J Crit Care Med, 2012; 16: 198 – 203.
- Otrock ZK and Eby CS. Clinical characteristics, prognostic factors,
and outcomes of adult patients with hemophagocytic lymphohistiocyosis. Am J Hematol, 2015; 3: 220 – 224. - Bae CB, Jung JY, Kim HA and Suh CH. Reactive hemophagocytic syndrome in adult-onset Still disease. Clinical feaures, predictive factors and prognosis in 21 patients. Medicine 2015, 94; 4: e451.
- Li F, Yang Y, Jin F, Dehoedt C, Rao J, Zhou Y, Li P, Yang G, Wang M, Zhang R and Yang Y. Clinical characteristics and prognostic factors of adult hemophagocytic syndrome patients: a retrospective study of increasing awareness of a disease from a single-center in China. Orphanet Journal of Rare Diseases 2015, feb 15; 10: 20.
- Parikh SA, Kapoor P, Letendre L, Kumar S and Wolanskyj AP. Prognostic factors and outcomeso adults with hemophagocytic lymphohistiocytosis. Mayo Clin Proc 2014 Apr, 89; 4: 484 – 492.
- Kaya Z, Bay A, Albayrak M, Kocak U, Yenicesu I and Gursel T. Prognostic Factors and Long-Term Outcome in 52 Turkish Children with Hemophagocytic Lymphohistiocytosis. Pediatr Crit Care Med 2015, 16; 6: e165 – e173.
- Buyse S, Teixeira L, Galicier L, Mariotte E, Lemiale V, Sequin A, Canet E, de Labarthe A, Darmon M, Rybojad M,Schelmmer B and Azoulay E. Critical care management of patients with hemophagocytic lymphohistiocytosis. Intensive Care Med 2010, 36; 10: 1695 – 1702.
- Yu JT, Wang CY, Yang Y, Wang RC, Chang KH, Hwuan WL and Teng CL. Lymphoma-associated hemophagocytic lymphohistiocytosis: experience in adults from a single institution. Ann Hematol 2013, 92; 11: 1529 – 1536.
- Demirkol D, Yildizdas D, Bayrakci B, Karapinar B, Kendirli T, Koroglu TF, Dursun O, Erkek N, Gedik H, Citak A, Kesici S, Karabocuoglu M and Carcillo JA. Hyperferritinemia in the critically ill child with secondary hemophagocytic lymphohistiocytosis / sepsis / multiple organ dysfunction syndrome / macrophage activation syndrome: what is the treatment? Critical care 2012; 16: R52.
- Nair V, Das S, Sharma A, Sharma S, Sharma P, Ray S and Bhattacharya S. A clinicopathological analysis of 26 patients with infection-associated haemophagocytic lymphohistiocytosis and the importance of bone marrow phagocytosis for the early initiation of immunomodulatory treatment. Postgrad Med J 2013; 89: 185 – 192.
- Trottestam H, Berglöf E, Horne AC, Onelöv E, Beutel K, Lehmberg K, Sieni E, Silfverberg T, Arico M, Janka G and Henter JI. Risk factors for early death in children with haemophagocytic Lymphohistiocytosis. Acta Pediatrica 2012; 101: 313 – 318.
- Shabbir M, Lucas J, Lazarchick J and Shirai K. Secondary hemophagocytic syndrome in adults: a case series of 18 patients in a single institution and a review of literature. Hematol Oncol 2011; 29: 100–106.
- Lin TF, Ferlic-Starck LL, Allen CE, Kozinetz CA and McClain KL. Rate of Decline of Ferritin in Patients with Hemophagocytic Lymphohistiocytosis as a Prognostic Variable for Mortality. Pediatr Blood Cancer 2011; 56: 154–155.
- Lu WX and Luo JM. Prognostic factors for hemophagocytic lymphohistiocytosis in children. Chin J Contemp Pediatr 2012, 14; 8: 593 – 597.